panGenomeBreedr_Workflows
Source:vignettes/panGenomeBreedr_Workflows.Rmd
panGenomeBreedr_Workflows.RmdTable of contents
Variant Discovery
Pangenome Data and Database Rationale
The examples used in this documentation are based on sorghum pangenome resources derived from whole-genome resequencing data of 1,676 sorghum lines. Variant calling was performed using version v5.1 of the BTx623 reference genome. The resulting SNP and INDEL variants were functionally annotated using snpEff.
Direct querying of snpEff-annotated VCF files from R is often computationally slow and inefficient, especially with large datasets. To overcome this limitation, we built a SQLite database that stores the variants, annotations, and genotypes in normalized tables. This structure allows for fast and flexible access to relevant data, supporting workflows for trait-predictive marker discovery.
We strongly recommend the creation of similar databases for other crops. The SQLite format offers a compact, portable, and queryable representation of pangenome-derived variant data, significantly improving performance and reproducibility in variant discovery pipelines.
A compressed format of the SQLite database for sorghum can be downloaded here.
Recommended Schema for the SQLite Database
The SQLite database contains the following three key tables:
variants
This table stores core metadata variant information extracted from the VCF.
| Column | Description |
|---|---|
variant_id |
Unique variant identifier |
chrom |
Chromosome name |
pos |
Genomic position (1-based) |
ref |
Reference allele |
alt |
Alternate allele |
variant_type |
Type of variant (e.g., SNP, indel) |
annotations
This table contains functional annotations from snpEff, typically including predicted effects, gene names, and functional categories.
| Column | Description |
|---|---|
variant_id |
Foreign key linking to variants
|
gene_name |
Sorghum gene ID (e.g., “Sobic.005G213600”) |
effect |
Type of effect (e.g., missense_variant) |
impact |
snpEff predicted impact (e.g., HIGH) |
feature_type |
Type of annotated feature (e.g., exon) |
| … | Additional snpEff annotation fields |
genotypes
This table stores genotype calls per sample for each variant in a wide format.
| Column | Description |
|---|---|
variant_id |
Foreign key linking to variants
|
chrom |
Chromosome name |
pos |
Genomic position |
sample1 |
Genotype for Sample 1 (e.g., 1|1) |
sample2 |
Genotype for Sample 2 (e.g., 0|0) |
| … | Genotypes for other samples |
Database Creation
We generated the SQLite database using a custom workflow that
- Parses a multi-sample VCF file annotated by snpEff,
- Extracts variant, annotation, and genotype data,
- Writes the data into normalized relational tables
(
variants,annotations,genotypes).
A prebuilt mini example database
(mini_sorghum_variant_vcf.db.gz) is included in the
extdata/ folder of the package.
Query Variant Tables
The query_db() function allows users to query specific
tables within a panGenomeBreedr-formatted SQLite database for variants,
annotations, or genotypes based on chromosome coordinates or candidate
gene IDs.
This function retrieves records from one of the following tables in the database:
-
variants: Basic variant information (chromosome, position, REF/ALT alleles, etc.) -
annotations: Variant effect predictions (e.g., from snpEff) -
genotypes: Genotypic data across lines/samples plus the metadata of the variants.
Users can specify genomic coordinates (chrom,
start, end) or a candidate gene name
(gene_name) to extract relevant entries.
If used correctly, the query_db() function returns a
data frame containing the filtered records from the selected table.
library(panGenomeBreedr)
# This example uses the mini SQLite database included in the package.
# Define a temporary path and decompress the example database
path <- tempdir()
mini_db <- system.file("extdata", "mini_sorghum_variant_vcf.db.gz",
package = "panGenomeBreedr", mustWork = TRUE)
mini_db_path <- file.path(path, 'mini_sorghum_variant_vcf.db')
R.utils::gunzip(mini_db, destname = mini_db_path, remove = FALSE)
# Query VCF genotypes within the genomic range: Chr05:75104537-75106403
gt_region <- query_db(db_path = mini_db_path,
chrom = "Chr05",
start = 75104537,
end = 75106403,
table_name = "genotypes")
# Query snpEff annotations within a candidate gene
annota_region <- query_db(db_path = mini_db_path,
chrom = "Chr05",
start = 75104537,
end = 75106403,
table_name = "annotations",
gene_name = "Sobic.005G213600")
# Clean up temporary files
unlink(list.files(tempdir(), full.names = TRUE, recursive = TRUE),
recursive = TRUE)| variant_id | chrom | pos | variant_type | ref | alt | IDMM | ISGC | ISGK | ISHC |
|---|---|---|---|---|---|---|---|---|---|
| INDEL_Chr05_75104541 | Chr05 | 75104541 | INDEL | T | TGAC | 0|0 | 0|0 | 0|0 | 0|0 |
| SNP_Chr05_75104557 | Chr05 | 75104557 | SNP | C | T | 0|0 | 0|0 | 0|0 | 0|0 |
| SNP_Chr05_75104560 | Chr05 | 75104560 | SNP | C | T | 0|0 | 0|0 | 0|0 | 0|0 |
| INDEL_Chr05_75104564 | Chr05 | 75104564 | INDEL | C | CA | 0|0 | 0|0 | 0|0 | 0|0 |
| SNP_Chr05_75104568 | Chr05 | 75104568 | SNP | G | T | 0|0 | 0|0 | 0|0 | 0|0 |
| variant_id | allele | annotation | impact | gene_name | gene_id | feature_type | feature_id | transcript_biotype | rank | HGVS_c | HGVS_p | chrom | pos | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | INDEL_Chr05_75104541 | TGAC | 3_prime_UTR_variant | MODIFIER | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.322_324dupGTC | Chr05 | 75104541 | |
| 4 | SNP_Chr05_75104557 | T | 3_prime_UTR_variant | MODIFIER | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.*309G>A | Chr05 | 75104557 | |
| 7 | SNP_Chr05_75104560 | T | 3_prime_UTR_variant | MODIFIER | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.*306G>A | Chr05 | 75104560 | |
| 10 | INDEL_Chr05_75104564 | CA | 3_prime_UTR_variant | MODIFIER | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.*301dupT | Chr05 | 75104564 | |
| 13 | SNP_Chr05_75104568 | T | 3_prime_UTR_variant | MODIFIER | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.*298C>A | Chr05 | 75104568 |
Summarize SnpEff Annotation and Impact
The query_ann_summary() function provides a convenient
way to summarize the distribution of SnpEff annotations
and impact categories across variant types (e.g., SNPs,
indels) within a defined genomic region.
This function enables users to quickly assess the types and functional implications of variants located within candidate genes or genomic intervals of interest.
library(panGenomeBreedr)
# Prepare test database
path <- tempdir()
mini_db <- system.file("extdata", "mini_sorghum_variant_vcf.db.gz",
package = "panGenomeBreedr", mustWork = TRUE)
mini_db_path <- file.path(path, 'mini_sorghum_variant_vcf.db')
R.utils::gunzip(mini_db, destname = mini_db_path, remove = FALSE)
# Run annotation summary for region Chr05:75104537-75106403
ann_summary <- query_ann_summary(db_path = mini_db_path,
chrom = "Chr05",
start = 75104537,
end = 75106403)
# Clean up
unlink(list.files(tempdir(), full.names = TRUE, recursive = TRUE),
recursive = TRUE)| annotation | variant_type | count | |
|---|---|---|---|
| 24 | upstream_gene_variant | SNP | 149 |
| 12 | upstream_gene_variant | INDEL | 53 |
| 19 | downstream_gene_variant | SNP | 46 |
| 22 | missense_variant | SNP | 29 |
| 23 | synonymous_variant | SNP | 21 |
| 13 | 3_prime_UTR_variant | SNP | 18 |
| impact | variant_type | count | |
|---|---|---|---|
| 8 | MODIFIER | SNP | 220 |
| 4 | MODIFIER | INDEL | 82 |
| 7 | MODERATE | SNP | 29 |
| 6 | LOW | SNP | 21 |
| 1 | HIGH | INDEL | 6 |
| 3 | MODERATE | INDEL | 5 |
The query_ann_summary() function returns a
list with the following elements:
annotation_summary: Data frame summarizing the count of each SnpEff annotation grouped by variant type.impact_summary: Data frame summarizing the count of each SnpEff impact level (e.g., HIGH, MODERATE) grouped by variant type.variant_type_totals: Total count of variants in the region grouped by variant type.
The annotation summary shows that there are six (6) INDEL
variants with a HIGH impact on protein function. To see these
variants, we need to use the query_by_impact() function, as
shown below:
# Prepare test database
path <- tempdir()
mini_db <- system.file("extdata", "mini_sorghum_variant_vcf.db.gz",
package = "panGenomeBreedr", mustWork = TRUE)
mini_db_path <- file.path(path, 'mini_sorghum_variant_vcf.db')
R.utils::gunzip(mini_db, destname = mini_db_path, remove = FALSE)
# Extract HIGH impact variant for a region or gene
high_variants <- query_by_impact(db_path = mini_db_path,
impact_level = 'high',
chrom = "Chr05",
start = 75104537,
end = 75106403)
# Clean up
unlink(list.files(tempdir(), full.names = TRUE, recursive = TRUE),
recursive = TRUE)| variant_id | chrom | pos | ref | alt | qual | filter | variant_type | allele | annotation | impact | gene_name | gene_id | feature_type | feature_id | transcript_biotype | rank | HGVS_c | HGVS_p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| INDEL_Chr05_75104881 | Chr05 | 75104881 | G | GTCGA | . | PASS | INDEL | GTCGA | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.1343_1344insTCGA | p.Ser449fs |
| INDEL_Chr05_75105587 | Chr05 | 75105587 | GC | G | . | PASS | INDEL | G | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.637delG | p.Ala213fs |
| INDEL_Chr05_75105598 | Chr05 | 75105598 | GT | G | . | PASS | INDEL | G | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.626delA | p.Asp209fs |
| INDEL_Chr05_75106156 | Chr05 | 75106156 | CGTAT | C | . | PASS | INDEL | C | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 2/2 | c.65_68delATAC | p.His22fs |
| INDEL_Chr05_75106295 | Chr05 | 75106295 | A | ATC | . | PASS | INDEL | ATC | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 1/2 | c.38_39dupGA | p.Ser14fs |
| INDEL_Chr05_75106325 | Chr05 | 75106325 | G | GTA | . | PASS | INDEL | GTA | frameshift_variant | HIGH | Sobic.005G213600 | Sobic.005G213600.v5.1 | transcript | Sobic.005G213600.1.v5.1 | protein_coding | 1/2 | c.8_9dupTA | p.Gln4fs |
Filter Variants by Allele Frequency
The query_by_af() and filter_by_af()
functions allow users to filter queried variants based on
alternate allele frequency thresholds within a
specified genomic region.
This is particularly useful for identifying polymorphic sites within candidate gene regions or windows of interest that meet desired minor allele frequency (MAF) thresholds for marker development.
An example usage for the filter_by_af() function is
shown in the code snippet below:
library(panGenomeBreedr)
# Define temporary directory and decompress demo database
path <- tempdir()
mini_db <- system.file("extdata", "mini_sorghum_variant_vcf.db.gz",
package = "panGenomeBreedr", mustWork = TRUE)
mini_db_path <- file.path(path, 'mini_sorghum_variant_vcf.db')
R.utils::gunzip(mini_db, destname = mini_db_path, remove = FALSE)
# Extract genotype data for all HIGH impact variants and filter by alternate allele frequency
geno_high_filtered <- query_genotypes(db_path = mini_db_path,
variant_ids = high_variants$variant_id,
meta_data = c('chrom', 'pos', 'ref', 'alt', 'variant_type')) |>
filter_by_af(min_af = 0.05)| variant_id | chrom | pos | ref_af | alt_af | |
|---|---|---|---|---|---|
| 1 | INDEL_Chr05_75104881 | Chr05 | 75104881 | 0.9495823 | 0.0504177 |
| 4 | INDEL_Chr05_75106156 | Chr05 | 75106156 | 0.9474940 | 0.0525060 |
| 5 | INDEL_Chr05_75106295 | Chr05 | 75106295 | 0.9439141 | 0.0560859 |
# Get genotype data for HIGH impact variants that passed allele filter
geno_high_filtered <- query_genotypes(db_path = mini_db_path,
variant_ids = geno_high_filtered$variant_id,
meta_data = c('chrom', 'pos', 'ref', 'alt', 'variant_type'))
# Clean up temporary files
unlink(list.files(tempdir(), full.names = TRUE, recursive = TRUE),
recursive = TRUE)KASP Marker Design
The kasp_marker_design() function enables the design of
KASP (Kompetitive Allele Specific PCR) markers from
identified putative causal variants. It supports SNP, insertion, and
deletion variants using VCF genotype data and a reference genome to
generate Intertek-compatible marker information, including upstream and
downstream polymorphic context.
This function automates the extraction of flanking sequences and polymorphic variants surrounding a focal variant and generates:
- Intertek-ready marker submission metadata
- DNA sequence alignment for visual inspection of marker context
- An optional publication-ready alignment plot in PDF format
The vcf file must contain the variant ID, Chromosome ID, Position, REF and ALT alleles, as well as the genotype data for samples, as shown in Table 1:
| variant_id | chrom | pos | ref | alt | variant_type | IDMM | ISGC |
|---|---|---|---|---|---|---|---|
| INDEL_Chr05_75104881 | Chr05 | 75104881 | G | GTCGA | INDEL | 0|0 | 0|0 |
| INDEL_Chr05_75106156 | Chr05 | 75106156 | CGTAT | C | INDEL | 0|0 | 0|0 |
| INDEL_Chr05_75106295 | Chr05 | 75106295 | A | ATC | INDEL | 0|0 | 0|0 |
# Example to design a KASP marker on a HIGH impact Deletion variant
library(panGenomeBreedr)
path <- tempdir() # (default directory for saving alignment outputs)
# Path to import sorghum genome sequence for Chromosome 5
path1 <- "https://raw.githubusercontent.com/awkena/panGB/main/Chr05.fa.gz"
# KASP marker design for variant ID: INDEL_Chr05_75106156 in Table 1
lgs1 <- kasp_marker_design(gt_df = geno_high_filtered,
variant_id_col = 'variant_id',
chrom_col = 'chrom',
pos_col = 'pos',
ref_al_col = 'ref',
alt_al_col = 'alt',
genome_file = path1,
geno_start = 7,
marker_ID = "INDEL_Chr05_75106156",
chr = "Chr05",
save_alignment = TRUE,
plot_file = path,
region_name = "lgs1")
# View marker alignment output from temp folder
path3 <- file.path(path, list.files(path = path, "alignment_"))
system(paste0('open "', path3, '"')) # Open PDF file from R
on.exit(unlink(path)) # Clear the temp directory on exitThe kasp_marker_design() function returns a list object
including a data.frame with marker design metadata:
-
SNP_Name: Variant ID -
SNP: Type of variant (SNP/INDEL) -
Marker_Name: Assigned name for the marker -
Chromosome: Chromosome name -
Chromosome_Position: Variant position -
Sequence: Intertek-style polymorphism sequence -
ReferenceAllele: Reference allele -
AlternativeAllele: Alternate allele
If save_alignment = TRUE, a PDF plot of
sequence alignment will be saved to plot_file.

The required sequence for submission to Intertek for the designed KASP marker is shown in Table 5.
| SNP_Name | SNP | Marker_Name | Chromosome | Chromosome_Position | Sequence | ReferenceAllele | AlternativeAllele |
|---|---|---|---|---|---|---|---|
| INDEL_Chr05_75106156 | Deletion | lgs1 | Chr05 | 75106156 | AACCACCGCCACTTCACCGCCGGCGCCGGGGACGACGGCTGCTGGCGCAGAGTAGCAAGTAGTAGTACTCATGCTGCTGCCTGCTGGTGTGTTTCGTATC[GTAT/-]GTAGGTGCCTATAAGAGTTTACATGCATGCATCAAAACTAAGGATCCCTCAATGTATGCATGACAGTTCAAAGAAGGCAACAAGGAAGAATTGCTTGCAT | GTAT |
|
KASP Marker Validation
The following example demonstrates how to use the customizable
functions in panGB to perform hypothesis testing of allelic
discrimination for KASP marker QC and validation.
panGB offers customizable functions for KASP marker
validation through hypothesis testing. These functions allow users to
easily perform the following tasks:
- Import raw or polished KASP genotyping results files (.csv) into
R.
Process imported data and assign FAM and HEX fluorescence colors for multiple plates.
Visualize marker QC using FAM and HEX fluorescence scores for each sample.
Validate the effectiveness of trait-predictive or background markers using positive controls.
Visualize plate design and randomization.
Reading Raw KASP Full Results Files (.csv)
The read_kasp_csv() function allows users to import raw
or polished KASP genotyping full results file (.csv) into R. The
function requires the path of the raw file and the row tags for the
different components of data in the raw file as arguments.
For polished files, the user must extract the Data
component of the full results file and save it as a csv file before
import.
By default, a typical unedited raw KASP data file uses the following
row tags for genotyping data: Statistics, DNA,
SNPs, Scaling, Data.
The raw file is imported as a list object in R. Thus, all components in the imported data can be extracted using the row tag ID as shown in the code snippet below:
# Import raw KASP genotyping file (.csv) using the read_kasp_csv() function
library(panGenomeBreedr)
# Set path to the directory where your data is located
# path1 <- "inst/extdata/Genotyping_141.010_01.csv"
path1 <- system.file("extdata", "Genotyping_141.010_01.csv",
package = "panGenomeBreedr",
mustWork = TRUE)
# Import raw data file
file1 <- read_kasp_csv(file = path1,
row_tags = c("Statistics", "DNA", "SNPs", "Scaling", "Data"),
data_type = 'raw')
# Get KASP genotyping data for plotting
kasp_dat <- file1$DataAssigning colors and PCH symbols for KASP cluster plotting
The next step after importing data is to assign FAM and HEX
fluorescence colors to samples based on their observed genotype calls.
This step is accomplished using the kasp_color() function
in panGB as shown in the code snippet below:
# Assign KASP fluorescence colors using the kasp_color() function
library(panGenomeBreedr)
# Create a subet variable called plates: masterplate x snpid
kasp_dat$plates <- paste0(kasp_dat$MasterPlate, '_',
kasp_dat$SNPID)
dat1 <- kasp_color(x = kasp_dat,
subset = 'plates',
sep = ':',
geno_call = 'Call',
uncallable = 'Uncallable',
unused = '?',
blank = 'NTC',
assign_cols = c(FAM = "blue", HEX = "gold" ,
het = "forestgreen"))The kasp_color() function requires the KASP genotype
call file as a data frame and can do bulk processing if there are
multiple master plates. The default values for the arguments in the
kasp_color() function are based on KASP annotations.
The kasp_color() function calls the
kasp_pch() function to automatically add PCH plotting
symbols that can equally be used to group genotypic clusters on the
plot.
When expected genotype calls are available for positive controls in KASP genotyping samples, we recommend the use of the PCH symbols for grouping observed genotypes instead of FAM and HEX colors.
The kasp_color() function expects that genotype calls
are for diploid state with alleles separated by a symbol. By default
KASP data are separated by : symbols.
The kasp_color() function returns a list object with the
processed data for each master plate as the components.
Cluster plot
To test the hypothesis that the designed KASP marker can accurately discriminate between homozygotes and heterozygotes (allelic discrimination), a cluster plot needs to be generated.
The kasp_qc_ggplot() and
kasp_qc_ggplot2()functions in panGB can be
used to make the cluster plots for each plate and KASP marker as shown
below:
# KASP QC plot for Plate 05
library(panGenomeBreedr)
kasp_qc_ggplot2(x = dat1[5],
pdf = FALSE,
Group_id = NULL,
scale = TRUE,
expand_axis = 0.6,
alpha = 0.9,
legend.pos.x = 0.6,
legend.pos.y = 0.75)
#> $`SE-24-1088_P01_d1_snpSB00804`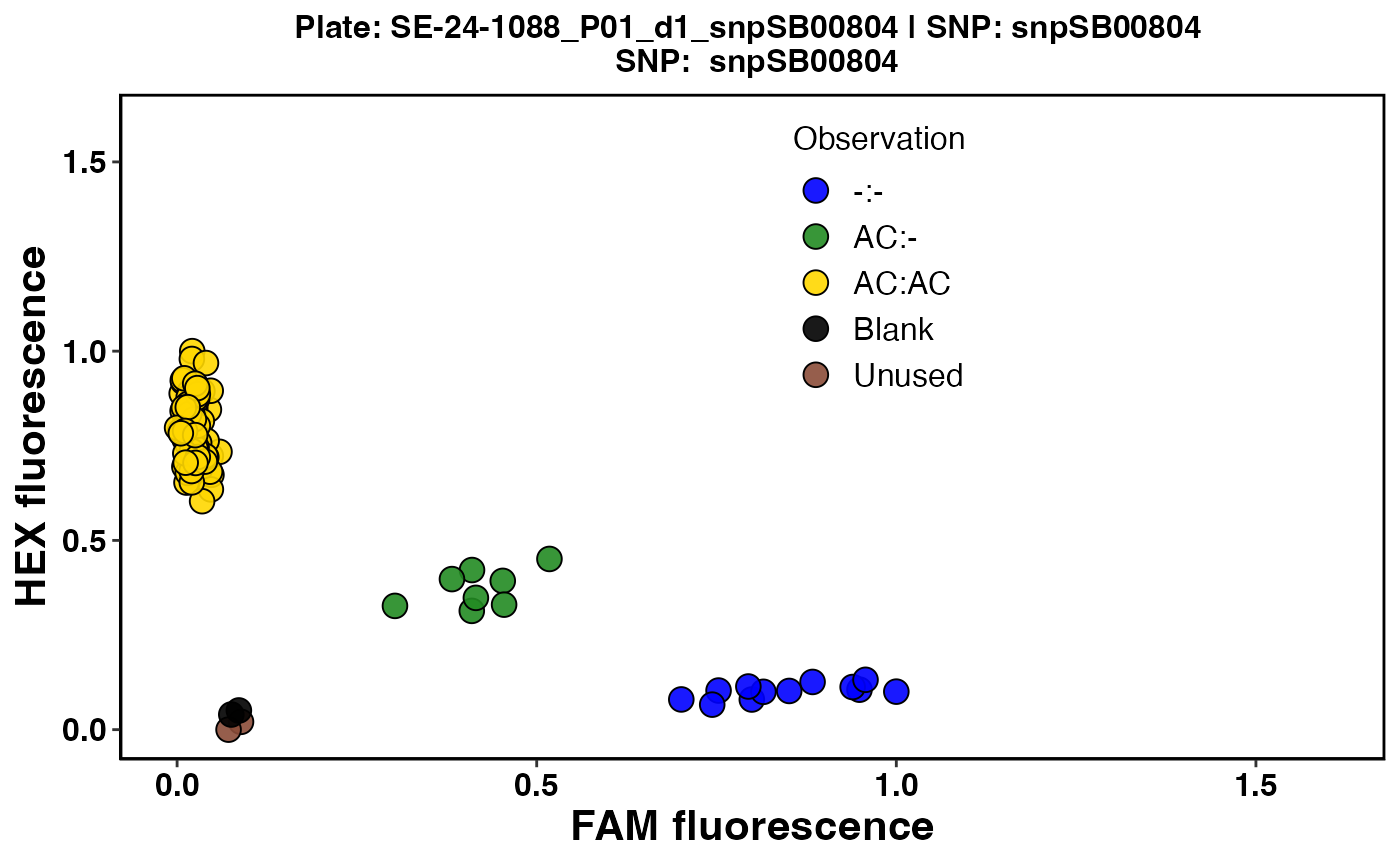
Fig. 3. Cluster plot for Plate 5 using FAM and HEX colors for grouping observed genotypes.
# KASP QC plot for Plate 05
library(panGenomeBreedr)
kasp_qc_ggplot2(x = dat1[5],
pdf = FALSE,
Group_id = 'Group',
Group_unknown = '?',
scale = TRUE,
pred_cols = c('Blank' = 'black', 'False' = 'firebrick3',
'True' = 'cornflowerblue', 'Unverified' = 'beige'),
expand_axis = 0.6,
alpha = 0.9,
legend.pos.x = 0.6,
legend.pos.y = 0.75)
#> $`SE-24-1088_P01_d1_snpSB00804`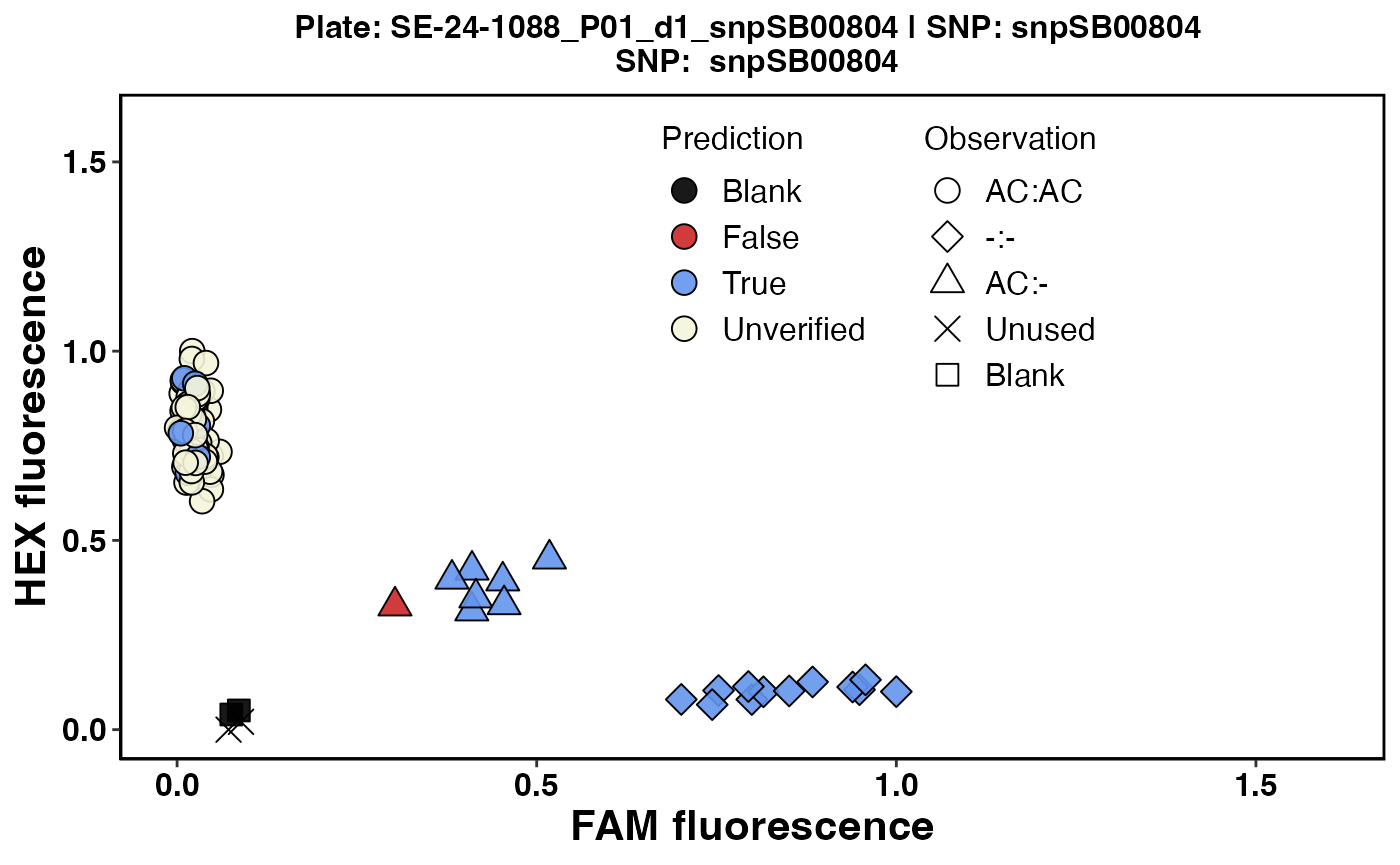
Fig. 4. Cluster plot for Plate 5 with an overlay of predictions for positive controls.
Color-blind-friendly color combinations are used to visualize verified genotype predictions (Figure 3).
In Figure 4, the three genotype classes are grouped based on plot PCH symbols using the FAM and HEX scores for observed genotype calls.
To simplify the verified prediction overlay for the expected genotypes for positive controls, all possible outcomes are divided into three categories (TRUE, FALSE, and UNVERIFIED) and color-coded to make it easier to visualize verified predictions.
BLUE (color code for the TRUE category) means genotype prediction matches the observed genotype call for the sample.
RED (color code for the FALSE category) means genotype prediction does not match the observed genotype call for the sample.
BEIGE (color code for the UNVERIFIED category) means three things: an expected genotype call could not be made before KASP genotyping, or an observed genotype call could not be made to verify the prediction.
Users can set the pdf = TRUE argument to save plots as a
PDF file in a directory outside R. The kasp_qc_ggplot() and
kasp_qc_ggplot2()functions can generate cluster plots for
multiple plates simultaneously.
To visualize predictions for positive controls to validate KASP
markers, the column name containing expected genotype calls must be
provided and passed to the function using the
Group_id = 'Group' argument as shown in the code snippets
above. If this information is not available, set the argument
Group_id = NULL.
Summary of Prediction Verification in Plates
The pred_summary() function produces a summary of
predicted genotypes for positive controls in each reaction plate after
verification (Table 3), as shown in the code snippet below:
# Get prediction summary for all plates
library(panGenomeBreedr)
my_sum <- pred_summary(x = dat1,
snp_id = 'SNPID',
Group_id = 'Group',
Group_unknown = '?',
geno_call = 'Call',
rate_out = TRUE)| plate | snp_id | false | true | unverified |
|---|---|---|---|---|
| SE-24-1088_P01_d1_snpSB00800 | snpSB00800 | 0.04 | 0.06 | 0.90 |
| SE-24-1088_P01_d2_snpSB00800 | snpSB00800 | 0.02 | 0.06 | 0.92 |
| SE-24-1088_P01_d1_snpSB00803 | snpSB00803 | 0.00 | 0.34 | 0.66 |
| SE-24-1088_P01_d2_snpSB00803 | snpSB00803 | 0.00 | 0.34 | 0.66 |
| SE-24-1088_P01_d1_snpSB00804 | snpSB00804 | 0.01 | 0.33 | 0.66 |
| SE-24-1088_P01_d2_snpSB00804 | snpSB00804 | 0.01 | 0.33 | 0.66 |
| SE-24-1088_P01_d1_snpSB00805 | snpSB00805 | 0.15 | 0.19 | 0.66 |
| SE-24-1088_P01_d2_snpSB00805 | snpSB00805 | 0.15 | 0.19 | 0.66 |
The output of the pred_summary() function can be
visualized as bar plots using the pred_summary_plot()
function as shown in the code snippet below:
# Get prediction summary for snp:snpSB00804
library(panGenomeBreedr)
my_sum <- my_sum$summ
my_sum <- my_sum[my_sum$snp_id == 'snpSB00804',]
pred_summary_plot(x = my_sum,
pdf = FALSE,
pred_cols = c('false' = 'firebrick3', 'true' = 'cornflowerblue',
'unverified' = 'beige'),
alpha = 1,
text_size = 12,
width = 6,
height = 6,
angle = 45)
#> $snpSB00804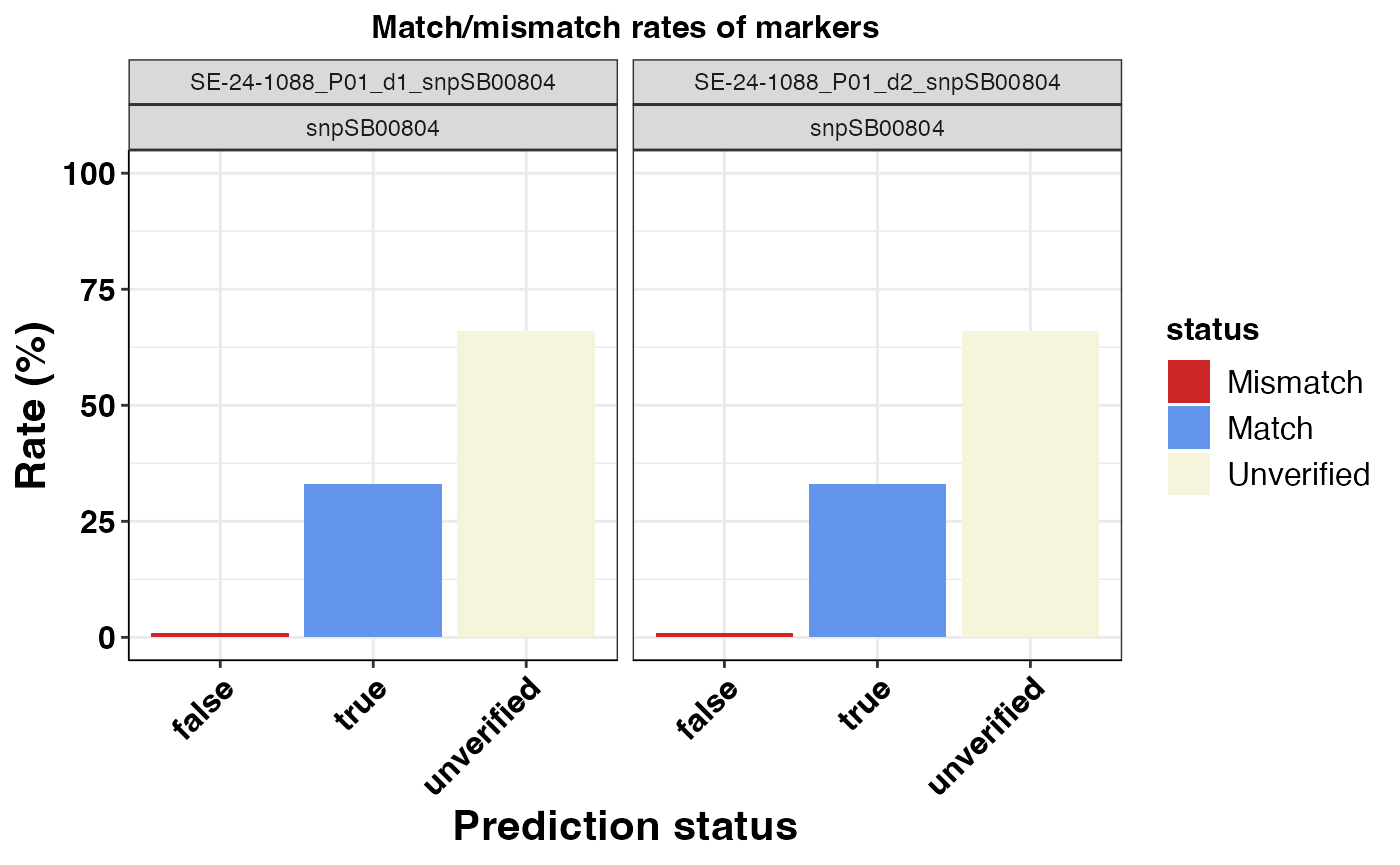
Fig. 5. Match/Mismatch rate of predictions for snp: snpSB00804.
Plot Plate Design
Users can visualize the observed genotype calls in a plate design
format using the plot_plate() function as depicted in
Figure 5, using the code snippet below:
plot_plate(dat1[5], pdf = FALSE)
#> $`SE-24-1088_P01_d1_snpSB00804`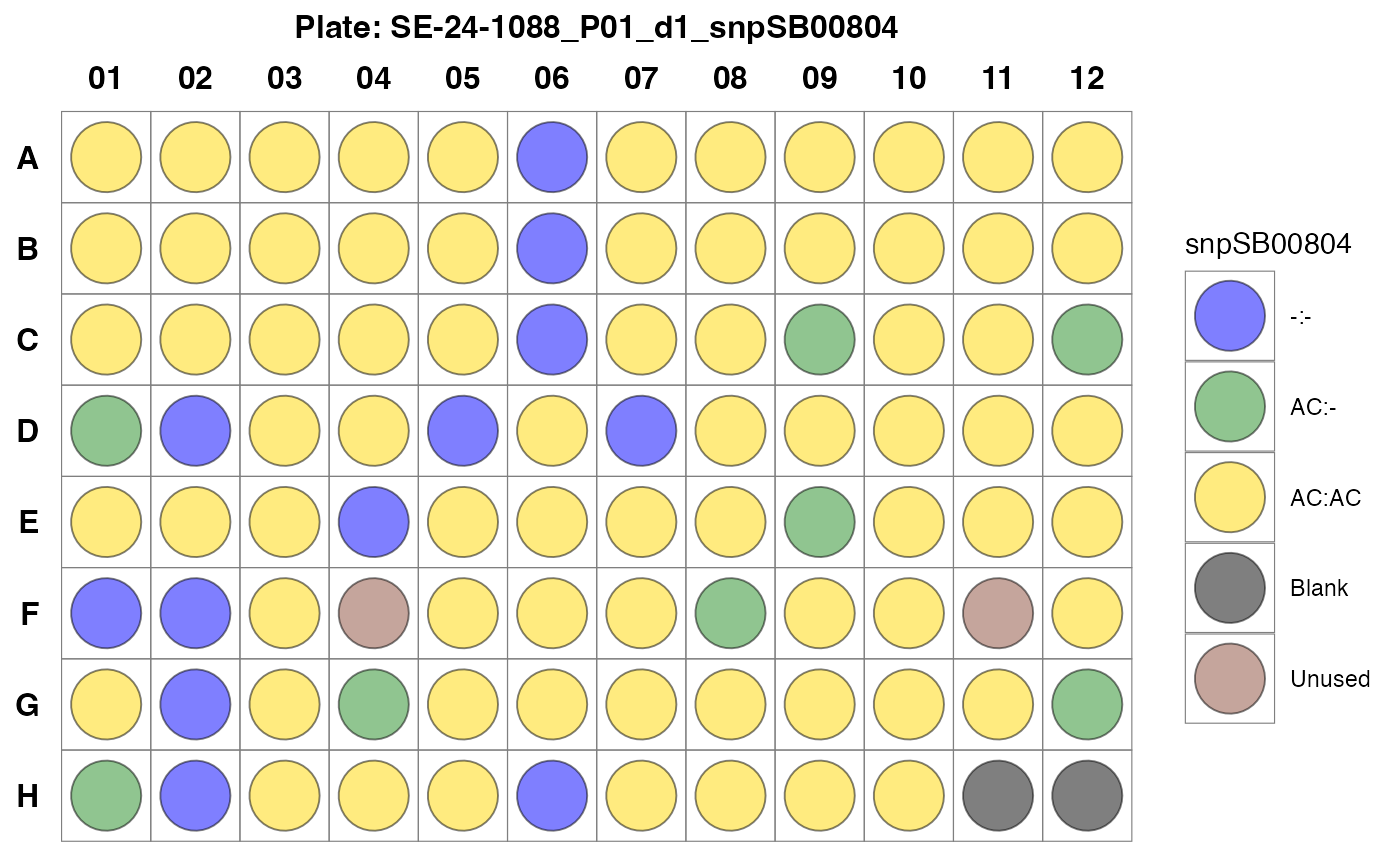
Fig. 6. Observed genotype calls for samples in Plate 5 in a plate design format.
Decision Support for Trait Introgression and MABC
panGB provides additional functionalities to test
hypotheses on the success of trait introgression pipelines and
crosses.
Users can easily generate heatmaps that compare the genetic background of parents to progenies to ascertain if a target locus was successfully introgressed or check for the hybridity of F1s. These plots also allow users to get a visual insight into the amount of parent germplasm recovered in progenies.
To produce these plots, users must have either polymorphic low or mid-density marker data and a map file for the markers. The map file must contain the marker IDs, their chromosome numbers and positions.
panGBcan handle data from KASP, Agriplex and DArTag
service providers.
Working with Agriplex Mid-Density Marker Data
Agriplex data is structurally different from KASP or DArTag data in
terms of genotype call coding and formatting. Agriplex uses
' / ' as a separator for genotype calls for heterozygotes,
and uses single nucleotides to represent homozygous SNP calls.
Creating Heatmaps with panGB
To exemplify the steps for creating heatmap, we will use a mid-density marker data for three groups of near-isogenic lines (NILs) and their parents (Table 4). The NILs and their parents were genotyped using the Agriplex platform. Each NIL group was genotyped using 2421 markers.
The imported data frame has the markers as columns and genotyped
samples as rows. It comes with some meta data about the samples. Marker
names are informative: chromosome number and position coordinates are
embedded in the marker names
(Eg. S1_778962: chr = 1, pos = 779862).
# Set path to the directory where your data is located
path1 <- system.file("extdata", "agriplex_dat.csv",
package = "panGenomeBreedr",
mustWork = TRUE)
# Import raw Agriplex data file
geno <- read.csv(file = path1, header = TRUE, colClasses = c("character")) # genotype calls
library(knitr)
knitr::kable(geno[1:6, 1:10], caption = 'Table 4: Agriplex data format', format = 'html', booktabs = TRUE)| Plate.name | Well | Sample_ID | Batch | Genotype | Status | S1_778962 | S1_1019896 | S1_1613105 | S1_1954298 |
|---|---|---|---|---|---|---|---|---|---|
| RHODES_PLATE1 | D04 | NIL_1 | 1 | RTx430a | Recurrent parent | A | G | G | A |
| RHODES_PLATE1 | F04 | NIL_2 | 1 | RTx430b | Recurrent parent | A | G | G | A |
| RHODES_PLATE1 | G04 | NIL_3 | 1 | IRAT204a | Donor parent | G | C | G | A |
| RHODES_PLATE1 | A05 | NIL_4 | 1 | IRAT204b | Donor Parent | G | C | G | A |
| RHODES_PLATE1 | D07 | NIL_5 | 1 | RMES1+|+_1 | NIL+ | A | G | G | A |
| RHODES_PLATE1 | F08 | NIL_6 | 1 | RMES1+|+_2 | NIL+ | A | G | G | A |
To create a heatmap that compares the genetic background of parents and NILs across all markers, we need to first process the raw Agriplex data into a numeric format. The panGB package has customizable data wrangling functions for KASP, Agriplex, and DArTag data.
The rm_mono() function can be used to filter out all
monomorphic loci from the data.
Since our imported Agriplex data has informative SNP IDs, we can use
the parse_marker_ns() function to generate a map file
(Table 5) for the markers.
The generated map file is then passed to the proc_kasp()
function to order the SNP markers according to their chromosome numbers
and positions.
The kasp_numeric() function converts the output of the
proc_kasp() function into a numeric format (Table 6). The
re-coding to numeric format is done as follows:
- Homozygous for Parent 1 allele = 1.
- Homozygous for Parent 2 allele = 0.
- Heterozygous = 0.5.
- Monomorphic loci = -1.
- Loci with a suspected genotype error = -2.
- Loci with at least one missing parental or any other genotype = -5.
# Parse snp ids to generate a map file
library(panGenomeBreedr)
# Data for stg5 NILs
stg5 <- geno[geno$Batch == 3, -c(1:6)]
rownames(stg5) <- geno$Genotype[17:25]
# Remove monomorphic loci from data
stg5 <- rm_mono(stg5)
# Parse snp ids to generate a map file
snps <- colnames(stg5) # Get snp ids
map_file <- parse_marker_ns(x = snps, sep = '_', prefix = 'S')
# order markers in map file
map_file <- order_markers(x = map_file)| snpid | chr | pos | |
|---|---|---|---|
| 1.317 | S1_402592 | 1 | 402592 |
| 1.1 | S1_778962 | 1 | 778962 |
| 1.633 | S1_825853 | 1 | 825853 |
| 1.318 | S1_1218846 | 1 | 1218846 |
| 1.2 | S1_1613105 | 1 | 1613105 |
| 1.319 | S1_1727150 | 1 | 1727150 |
| 1.3 | S1_1954298 | 1 | 1954298 |
| 1.4 | S1_1985365 | 1 | 1985365 |
# Process genotype data to re-order SNPs based on chromosome and positions
stg5 <- proc_kasp(x = stg5,
kasp_map = map_file,
map_snp_id = "snpid",
sample_id = "Genotype",
marker_start = 1,
chr = 'chr',
chr_pos = 'pos')
# Convert to numeric format for plotting
num_geno <- kasp_numeric(x = stg5,
rp_row = 1,
dp_row = 3,
sep = ' / ',
data_type = 'agriplex')| S1_402592 | S1_778962 | S1_825853 | S1_1218846 | S1_1613105 | S1_1727150 | S1_1954298 | S1_1985365 | |
|---|---|---|---|---|---|---|---|---|
| BTx623a | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| BTx623b | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| BTx642a | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BTx642b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Stg5+|+_1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Stg5+|+_2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 |
| Stg5-|-_1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Stg5-|-_2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Stg5-|-_3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
All is now set to generate the heatmap (Figure 6) using the
cross_qc_ggplot() function, as shown in the code snippet
below:
# Get prediction summary for snp:snpSB00804
library(panGenomeBreedr)
# Create a heatmap that compares the parents to progenies
cross_qc_heatmap(x = num_geno,
map_file = map_file,
snp_ids = 'snpid',
chr = 'chr',
chr_pos = 'pos',
parents = c("BTx623a", "BTx642a"),
pdf = FALSE,
filename = 'background_heatmap',
legend_title = 'stg5_NILs',
alpha = 0.9,
text_size = 15)
#> $Batch1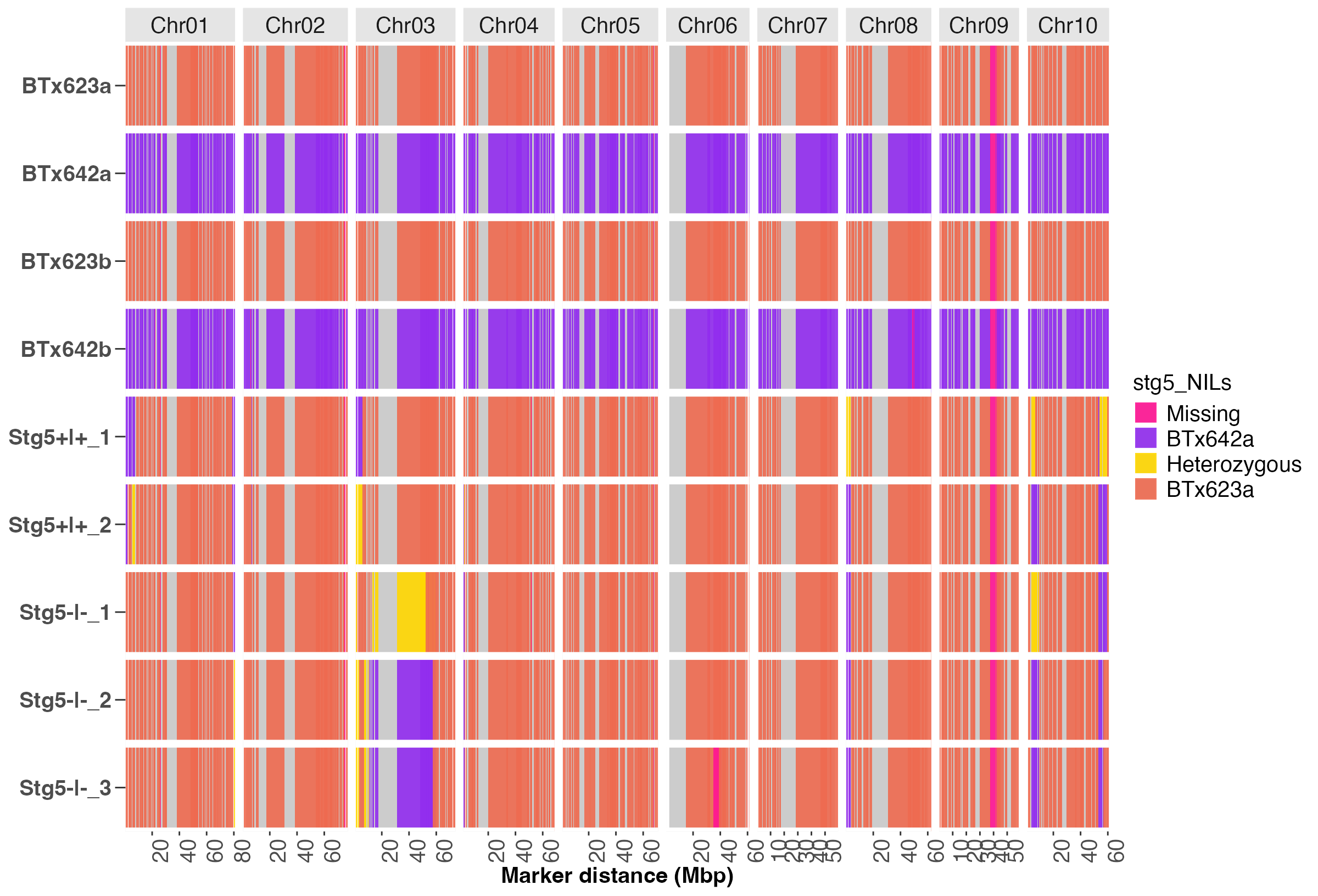
Fig. 6. A heatmap that compares the genetic background of parents and stg5 NIL progenies across all markers.
The cross_qc_ggplot() function is a wrapper for
functions in the ggplot2 package.
Users must specify the IDs for the two parents using the
parents argument. In the code snippet above, the recurrent
parent is BTx623 and the donor parent for the stg5
locus is BTx642.
The group_sz argument must be specified to plot the
heatmap in batches of progenies to avoid cluttering the plot with many
observations.
Users can set the pdf = TRUE argument to save plots as a
PDF file in a directory outside R.
Trait Introgression Hypothesis Testing
To test the hypothesis that the stg5 NIL development was
effective, we can use the cross_qc_annotate() function to
generate a heatmap (Figure 7) with an annotation of the position of the
stg5 locus on Chr 1, as shown below:
###########################################################################
# Subset data for the first 30 markers on Chr 1
stg5_ch1 <- num_geno[, map_file$chr == 1][,1:20]
# Get the map file for subset data
stg5_ch1_map <- parse_marker_ns(colnames(stg5_ch1))
# Annotate a heatmap to show the stg5 locus on Chr 1
# The locus is between positions 1e6 - 1.4 Mbp on Chr 1
cross_qc_heatmap(x = stg5_ch1,
map_file = stg5_ch1_map,
snp_ids = 'snpid',
chr = 'chr',
chr_pos = 'pos',
parents = c("BTx623a", "BTx642a"),
trait_pos = list(stg5 = c(chr = 1, start = 1e6, end = 1.4e6)),
text_scale_fct = 0.18,
pdf = FALSE,
legend_title = 'Stg5_NILs',
alpha = 0.9,
text_size = 15)
#> $Batch1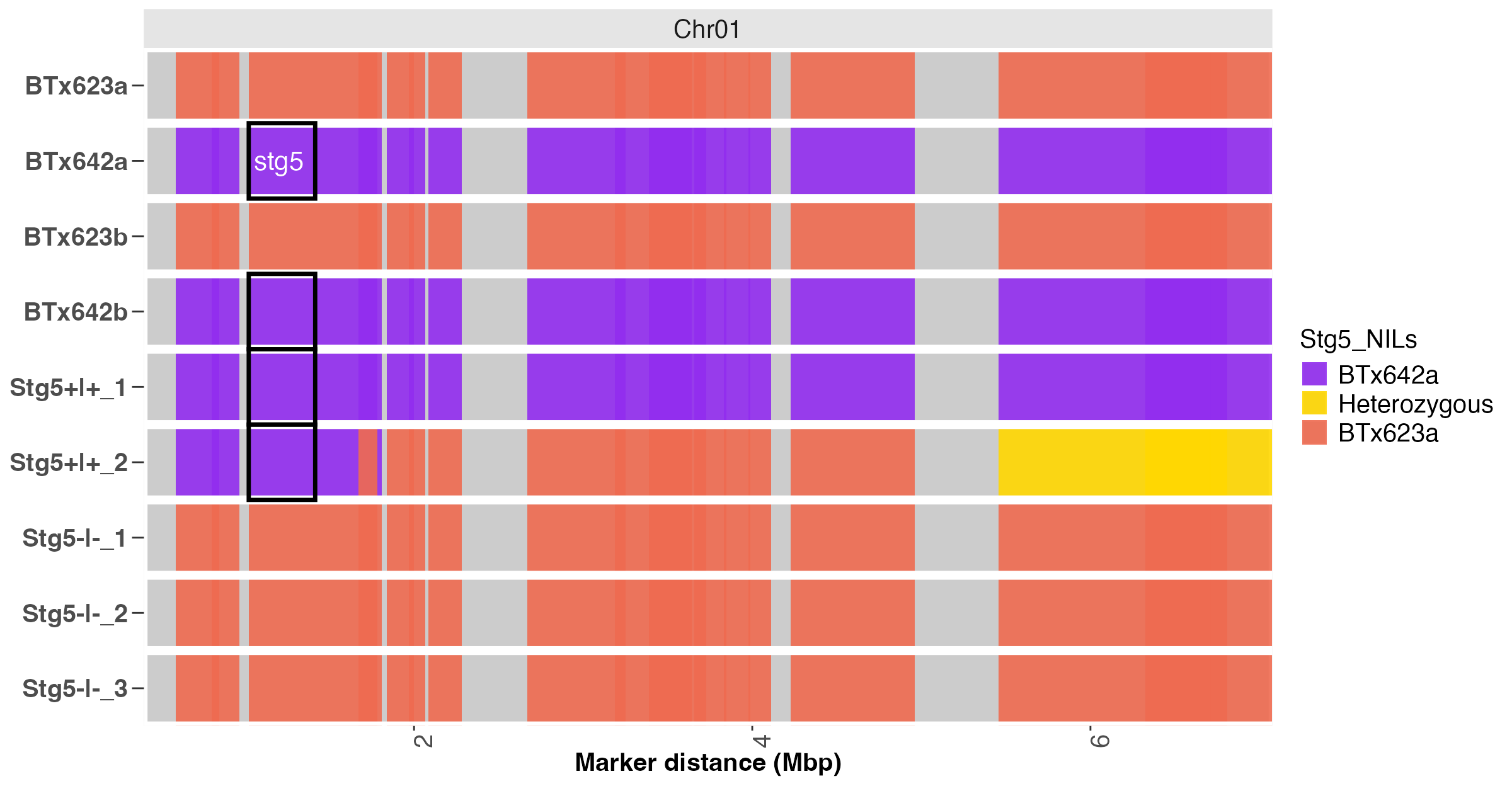
Fig. 7. Heatmap annotation of the stg5 locus on Chr 1.
In the code snippet above, the numeric matrix of genotype calls and its associated map file are required.
The recurrent and donor parents must be specified using the
parents argument.
The snp_ids, chr, and chr_pos arguments can be used to
specify the column names for marker IDs, chromosome number and positions
in the attached map file.
The trait_pos argument was used to specify the position of
the target locus (stg5) on chromosome one. Users can specify
the positions of multiple target loci as components of a list object for
annotation.
In Figure 7, the color intensity correlates positively with the marker density or coverage. Thus, areas with no color (white vertical gaps) depicts gaps in the marker coverage in the data.
Decision Support for MABC
Users can use the calc_rpp_bc() function in
panGB to calculate the proportion of recurrent parent
background (RPP) fully recovered in backcross progenies.
It also returns the rpp value for each chromosome.
In the computation, partially regions are ignored, hence, heterozygous scores are not used.
The output for he calc_rpp_bc() function can be passed
to the rpp_barplot() function to visualize the computed RPP
values for progenies as a bar plot. Users can specify an RPP threshold
to easily identify lines that have RPP values above or equal to the
defined RPP threshold on the bar plot.
We can compute and visualize the observed RPP values for the stg5 NILs across all polymorphic loci as shown in the code snippet below:
# Calculate weighted RPP
rpp <- calc_rpp_bc(x = num_geno,
map_file = map_file,
map_chr = 'chr',
map_pos = 'pos',
map_snp_ids = 'snpid',
rp = 1,
rp_num_code = 1,
na_code = -5,
weighted = TRUE)
# Generate bar plot for RPP values
rpp_barplot(rpp_df = rpp,
rpp_threshold = 0.93,
text_size = 18,
text_scale_fct = 0.1,
alpha = 0.9,
bar_width = 0.5,
aspect_ratio = 0.5,
pdf = FALSE)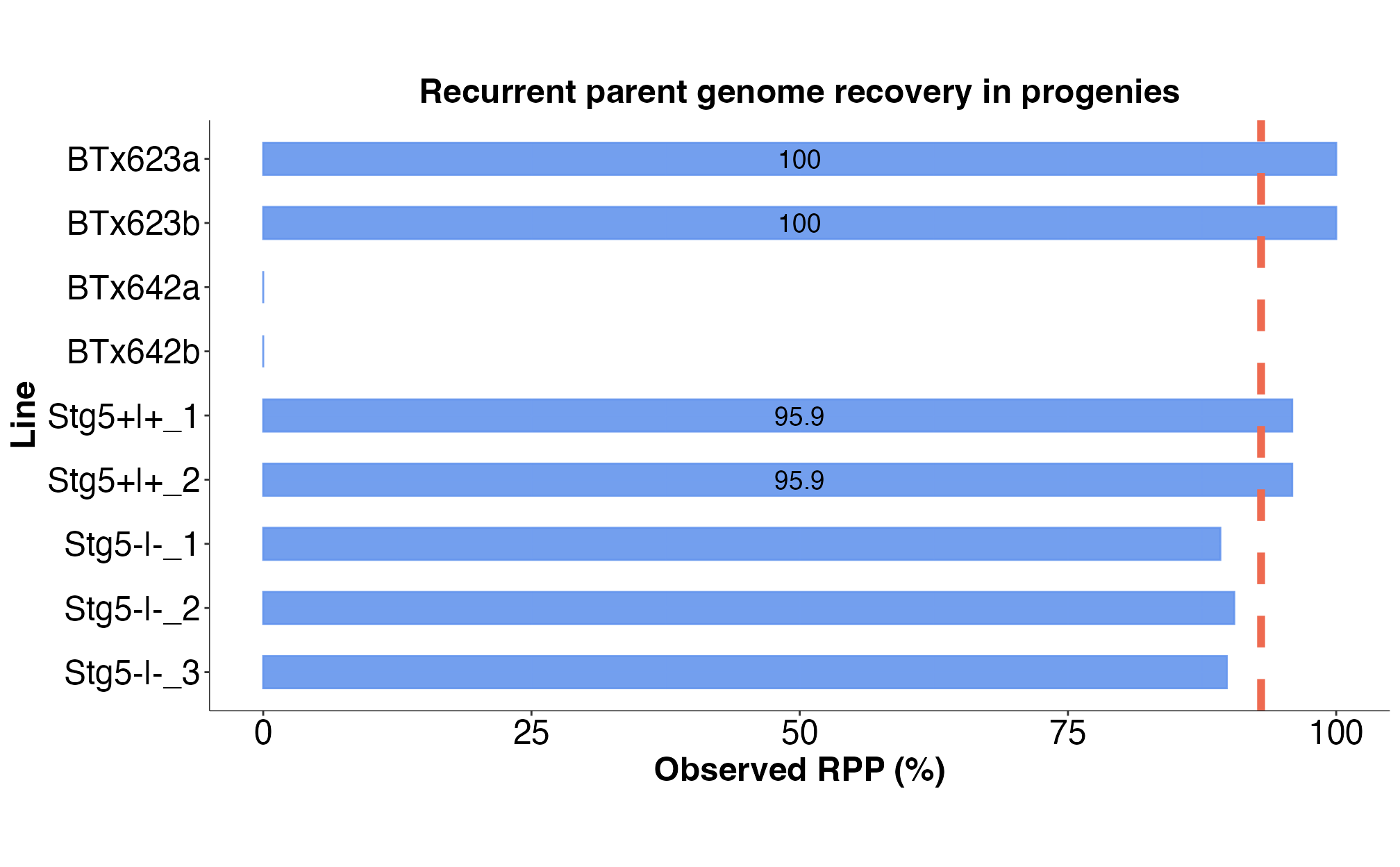
Fig. 8. Computed RPP values for the stg5 NILs.
| sample_id | chr_1 | chr_2 | chr_3 | chr_4 | chr_5 | chr_6 | chr_7 | chr_8 | chr_9 | chr_10 | total_rpp | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BTx623a | BTx623a | 1.000 | 1.000 | 1.000 | 1.000 | 1 | 1.000 | 1 | 1.000 | 1 | 1.000 | 1.000 |
| BTx623b | BTx623b | 1.000 | 1.000 | 1.000 | 1.000 | 1 | 1.000 | 1 | 1.000 | 1 | 1.000 | 1.000 |
| BTx642a | BTx642a | 0.000 | 0.000 | 0.000 | 0.000 | 0 | 0.000 | 0 | 0.000 | 0 | 0.000 | 0.000 |
| BTx642b | BTx642b | 0.000 | 0.000 | 0.000 | 0.000 | 0 | 0.000 | 0 | 0.000 | 0 | 0.000 | 0.000 |
| Stg5+|+_1 | Stg5+|+_1 | 0.880 | 0.997 | 0.931 | 0.995 | 1 | 1.000 | 1 | 0.949 | 1 | 0.840 | 0.959 |
| Stg5+|+_2 | Stg5+|+_2 | 0.924 | 0.997 | 0.939 | 0.987 | 1 | 1.000 | 1 | 0.949 | 1 | 0.796 | 0.959 |
| Stg5-|-_1 | Stg5-|-_1 | 0.982 | 1.000 | 0.455 | 0.984 | 1 | 0.759 | 1 | 0.949 | 1 | 0.796 | 0.892 |
| Stg5-|-_2 | Stg5-|-_2 | 0.991 | 1.000 | 0.286 | 0.987 | 1 | 0.995 | 1 | 0.949 | 1 | 0.845 | 0.905 |
| Stg5-|-_3 | Stg5-|-_3 | 0.991 | 1.000 | 0.278 | 0.987 | 1 | 0.931 | 1 | 0.949 | 1 | 0.845 | 0.898 |
The calc_rpp_bc() function in panGB
provides two algorithms for computing the observed RPP values: weighted
and unweighted RPP values. We recommend the use of the weighted
algorithm to account for differences in the marker coverage across the
genome.
The algorithm for the weighted RPP values is explained below.
Weighted RPP Computation in panGB
Let represent the weight for marker , based on the relative distances to its adjacent markers.
For a set of markers with positions , where represents the distance between adjacent markers, the weights can be calculated as follows:
-
For the first marker :
-
For a middle marker :
-
For the last marker :
where:
- is the distance between marker and marker ,
- is the total distance across all segments, used for normalization.
Let represent the Recurrent Parent Proportion based on relative distance weighting. If is the weight for each marker , and represents whether marker matches the recurrent parent if it matches, otherwise), then the weighted RPP is calculated as:
The unweighted RPP is calculated without the use of the weights as follows:
where:
- is the weight of marker , calculated based on the relative distance it covers,
- is the match indicator for marker (1 if matching the recurrent parent, 0 otherwise),
- is the total number of markers.
This formula provides the sum of the weighted contributions from each marker, representing the proportion of the recurrent parent genome in the individual.
Decision Support for Foreground Selection
The foreground_select() and find_lines()
functions are designed to help breeders identify lines that carry
favorable alleles at target loci using trait-predictive
markers. This process supports foreground selection in
marker-assisted selection pipelines.
Generate a Binary Matrix: foreground_select()
function
The foreground_select() function score Lines for
presence of favorable alleles by converting raw marker genotype data
into a binary matrix (1 = favorable allele present, 0 = absent) based on
a set of trait-predictive markers. The foreground_select()
function is shown in the code snippet below:
library(panGenomeBreedr)
# Marker genotype data
geno <- data.frame(SNP1 = c("A:A", "A:G", "G:G", "A:A"),
SNP2 = c("C:C", "C:T", "T:T", "C:T"),
SNP3 = c("G:G", "G:G", "A:G", "A:A"),
row.names = c("Line1", "Line2", "Line3", "Line4"))
# Trait-predictive marker metadata
marker_info <- data.frame(qtl_markers = paste0("SNP", 1:3),
locus_name = paste0('loc', 1:3),
fav_alleles = c("A", "C", "G"),
alt_alleles = c("G", "T", "A"))
# Convert raw genotypes to binary (foreground profile)
foreground_matrix <- foreground_select(geno_data = geno,
fore_marker_info = marker_info,
fore_marker_col = "qtl_markers",
fav_allele_col = "fav_alleles",
alt_allele_col = "alt_alleles",
select_type = "homo")| SNP1 | SNP2 | SNP3 | |
|---|---|---|---|
| Line1 | 1 | 1 | 1 |
| Line2 | 0 | 0 | 1 |
| Line3 | 0 | 0 | 0 |
| Line4 | 1 | 0 | 0 |
The foreground_select() function has the following input
parameters:
| Argument | Type | Description |
|---|---|---|
geno_data |
data.frame |
Marker genotype data (lines x markers), e.g., "A:A",
"A:G"
|
fore_marker_info |
data.frame |
Metadata describing trait-predictive markers, including marker names, favorable alleles, and alternate alleles |
fore_marker_col |
character |
Column name in fore_marker_info for marker names |
fav_allele_col |
character |
Column name for favorable allele |
alt_allele_col |
character |
Column name for alternate allele |
select_type |
character |
Genotype class to select: "homo",
"hetero", or "both"
|
sep |
character |
Separator used in genotype calls (e.g., ":") |
Visualize the Binary Data with UpSet Plot
Generate an UpSet plot using the UpSetR package to
explore the co-occurrence of favorable alleles across lines. The UpSet
plot allows users to quickly determine:
Whether any line carries favorable alleles at all target loci.
How favorable alleles are distributed across lines.
Which loci are rarely combined.
How to Interpret the UpSet Plot:
Top bar plot: shows the number of lines for each unique combination (intersection) of target loci with favorable alleles.
Bottom matrix of dots and lines: indicates which loci are involved in each combination.
Left bar plot: shows how many lines have the favorable allele for each individual target locus.
# Make an Upset plot and overlay with trait loci names
metadata <- data.frame(sets = marker_info$qtl_markers,
locus = marker_info$locus_name)
nl <- ncol(foreground_matrix) # Number of markers
UpSetR::upset(foreground_matrix,
nsets = nl,
mainbar.y.label = "Locus Intersection Size",
sets.x.label = "Locus Size",
text.scale = 1.2,
set.metadata = list(data = metadata,
plots = list(list(type = 'text',
assign = 8,
column = 'locus',
colors = rep('firebrick2', nl)))))
#> Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
#> ℹ Please use tidy evaluation idioms with `aes()`.
#> ℹ See also `vignette("ggplot2-in-packages")` for more information.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue to the authors.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue to the authors.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue to the authors.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.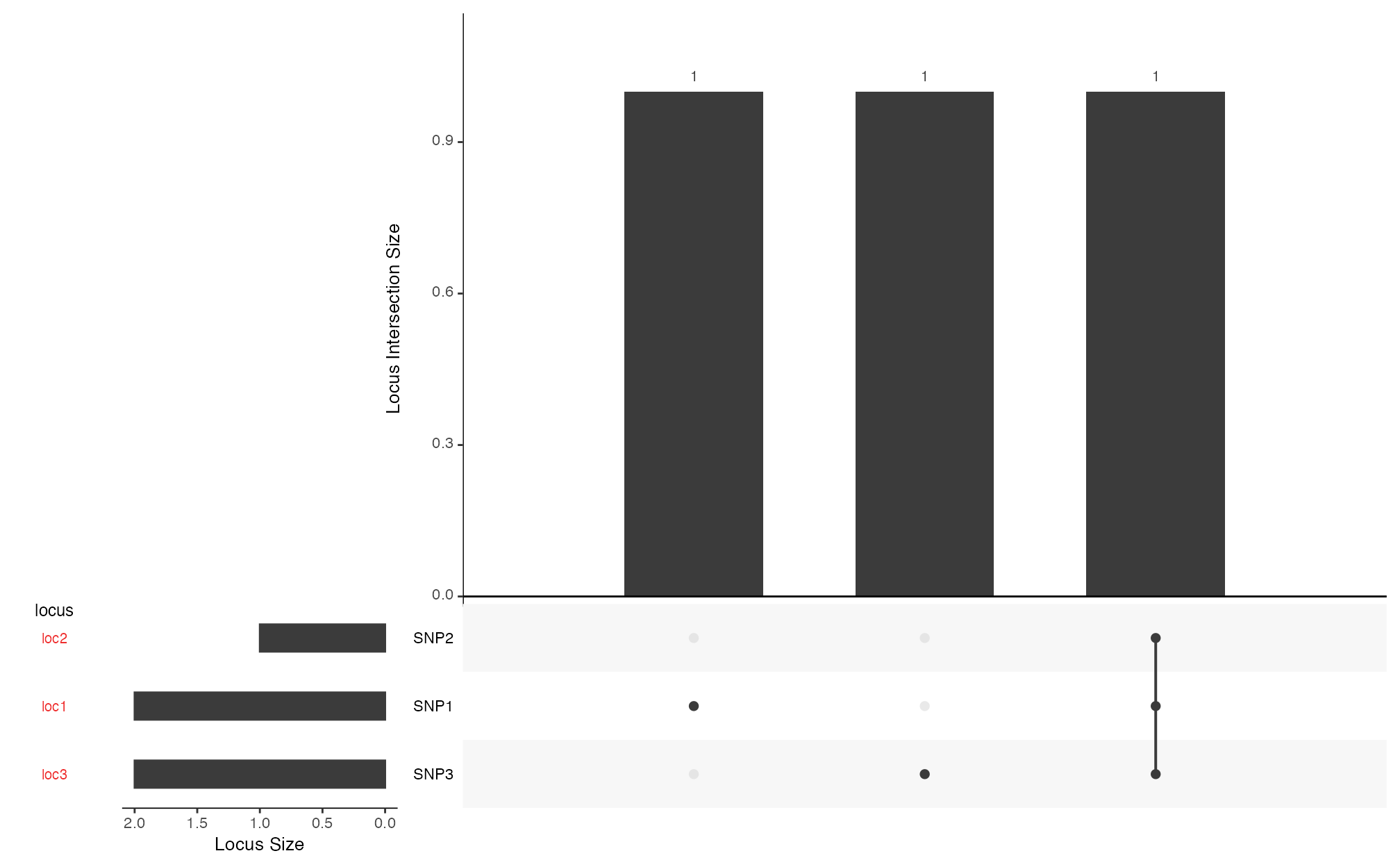
Fig. 9. Visualizing the co-occurrence of favorable alleles across lines.
Query Lines by Intersection Category: find_lines()
function
The find_lines() function identifies lines based on
target loci profile by filtering the binary output of
foreground_select() to return line names that match a
desired allele presence/absence profile across loci. Using the binary
matrix, users can extract line IDs corresponding to any intersection
(i.e., specific combinations of favorable alleles revealed by the UpSet
plot).
library(panGenomeBreedr)
# Find lines with favorable alleles at all target loci
selected_lines <- find_lines(mat = foreground_matrix,
present = c('SNP1', 'SNP2', 'SNP3'))
print(selected_lines)
#> [1] "Line1"The find_lines() function has the following input
parameters:
| Argument | Type | Description |
|---|---|---|
mat |
data.frame |
Binary matrix. |
present |
character |
Markers that must be 1. |
absent |
character |
Markers that must be 0. |
Troubleshooting
If the package does not run as expected, check the following:
Was the package properly installed?
Do you have the required dependencies installed?
Were any warnings or error messages returned during package installation?
Are all packages up to date before installing panGB?
Support and Feedback
For support and submission of feedback, email the maintainer Alexander Kena, PhD at alex.kena24@gmail.com